Komplexen Moleküldynamiken auf der Spur
Neuer Algorithmus treibt den Einsatz von KI in den Materialwissenschaften voran.
Forschern der TU Berlin ist es in Zusammenarbeit mit Google Research gelungen, einen Algorithmus zu entwickeln, der anhand von quantenmechanischen Daten den potenziellen Energiezustand von einzelnen Molekülen mit großer Genauigkeit und Effizienz vorhersagen kann. Damit könnten sich speziell für Materialwissenschaftler ganz neue Optionen ergeben.
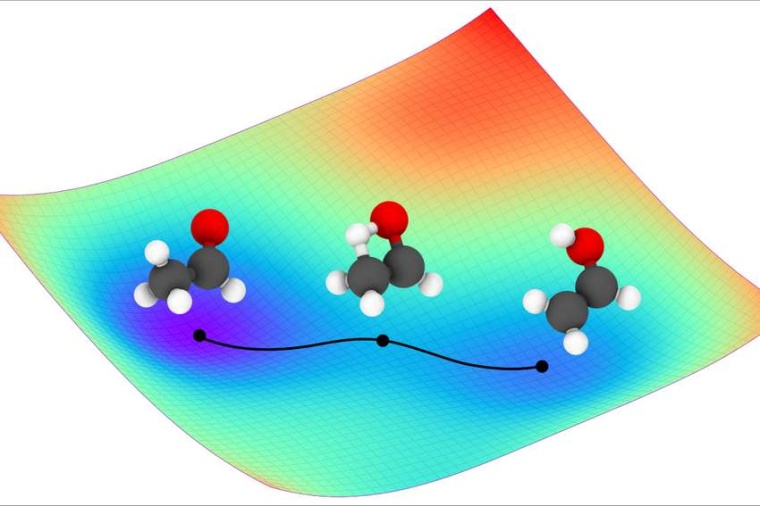
„Quantenmechanik befasst sich unter anderem mit den chemischen und physikalischen Eigenschaften eines Moleküls auf Basis der räumlichen Anordnung ihrer Atome. Eine chemische Reaktion wiederum beruht auf dem Zusammenspiel vieler Moleküle und ist ein multidimensionaler Prozess“, erläutert Klaus-Robert Müller von der TU Berlin. Die einzelnen Schritte einer chemischen Reaktion auf Molekülebene oder sogar auf atomarer Ebene vorherzusagen und zu modellieren ist ein lang gehegter Traum vieler Materialwissenschaftler.
Eine entscheidende Rolle für die Reaktionsfähigkeit von Molekülen spielt die Potenzialhyperfläche. Sie beschreibt die Abhängigkeit der Energie der Atome eines Moleküls von der geometrischen Anordnung der Atomkerne. Die Kenntnis der ultragenauen Potenzialhyperflächen von Molekülen erlaubt es, die Bewegung einzelner Atome, etwa während einer chemischen Reaktion, zu simulieren, um deren dynamische quantenmechanische Eigenschaften besser zu verstehen und dadurch Ablauf und Ergebnis von Reaktionen exakt vorherzusagen.
„Man kann sich die Potenzialhyperfläche wie eine Landschaft mit Bergen und Tälern vorstellen. Ähnlich wie bei einer Murmel, die über eine Miniaturversion dieser Landschaft rollt, wird die Bewegung der Atome durch die Berge und Täler auf der Potenzialhyperfläche bestimmt: Man nennt das auch Molekulardynamik“, erklärt Oliver Unke von Google Research in Berlin.
Im Gegensatz zu vielen anderen Anwendungsgebieten des maschinellen Lernens, in denen einer KI häufig nahezu endlose Datenmengen zur Verfügung stehen, sind für die Vorhersage von Potenzialhyperflächen typischerweise nur wenige quantenmechanische Referenzdaten vorhanden, die unter Einsatz von enormer Rechenleistung erzeugt werden müssen.
„So kann die exakte mathematische Modellierung molekulardynamischer Eigenschaften zwar einerseits teure und zeitaufwändige Laborexperimente einsparen, benötigt aber im Gegenzug unverhältnismäßig hohe Rechenleistungen. Wir hoffen, dass unser neuartiger Deep Learning Algorithmus – ein Transformer Modell, das erstmals auch Spin und Ladung von Atomen berücksichtigt – zu neuen Erkenntnissen im Bereich der Chemie, Biologie und Materialwissenschaften führen wird – bei deutlich geringerer Rechenleistung“, so Müller.
Um eine besonders hohe Daten-Effizienz zu erreichen, kombiniert das neue Deep Learning Modell der Forscher die KI mit bekannten physikalischen Gesetzen. Bestimmte Aspekte der Potenzialhyperfläche können mit einfachen physikalischen Formeln sehr genau beschrieben werden. Die neue Methode erlernt daher nur die Anteile der Potenzialhyperfläche, für die keine einfache mathematische Beschreibung verfügbar ist. „Sehr praktisch: Die KI muss nur das lernen, was man noch nicht aus der Physik weiß“, so Müller. Dadurch kann Rechenleistung eingespart werden.
Eine weitere Besonderheit ist, dass der Algorithmus auch nichtlokale Wechselwirkungen beschreiben kann. Nichtlokalität bedeutet in diesem Zusammenhang, dass eine Veränderung an einem Atom an einer bestimmten geometrischen Position des Moleküls Einfluss auf Atome an einer räumlich getrennten geometrischen Molekülposition haben kann. Aufgrund der räumlichen Trennung von Ursache und Wirkung sind diese Eigenschaften von Quantensystemen besonders schwer für eine KI zu lernen.
Die Forscher lösten dieses Problem mit einem Transformer, einer Methode, welche ursprünglich für die maschinelle Verarbeitung von Sprache und Texten oder auch Bildern entwickelt wurde. „In einem Text ist die Bedeutung eines Wortes oder Satzes häufig stark vom Kontext abhängig. Dabei kann die relevante Kontext-Information in einem völlig anderen Textabschnitt stehen. In diesem Sinne ist auch Sprache auf eine Art und Weise nichtlokal“, erklärt Müller den Zusammenhang.
Mit Hilfe eines solchen Transformers können die Wissenschaftler auch verschiedene elektronische Zustände eines Moleküls wie Spin und Ladung unterscheiden. „Das ist zum Beispiel relevant für physikalische Prozesse in Solarzellen, in denen ein Molekül Licht absorbiert und dadurch in einen anderen elektronischen Zustand versetzt wird“, so Unke.
TU Berlin / RK
Weitere Infos
- Originalveröffentlichung
O. T. Unke et al.: SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects, Nat. Commun. 12, 7273 (2021); DOI: 10.1038/s41467-021-27504-0 - Berlin Institute for the Foundation of Learning and Data, Technische Universität Berlin
- Brain Team, Google Research, Berlin










